


HEC Biomolecular Simulation Consortium (HECBioSim)
The HEC BioSim consortium focusses on molecular simulations, at a variety of time and length scales but based on well-defined physics to complement experiment. The unique insight they can provide gives molecular level understanding of how biological macromolecules function. Simulations are crucial in analysing protein folding, mechanisms of biological catalysis, and how membrane proteins interact with lipid bilayers. A particular challenge is the integration of simulations across length and timescales: different types of simulation method are required for different types of problems.
| Website | http://www.hecbiosim.ac.uk/ |
| Contact | Contact (James Gebbie) |
| Consortium Head | Prof. Syma Khalid |
| ARCHER CSE Consortium Contact | Dr Neelofer Banglawala |
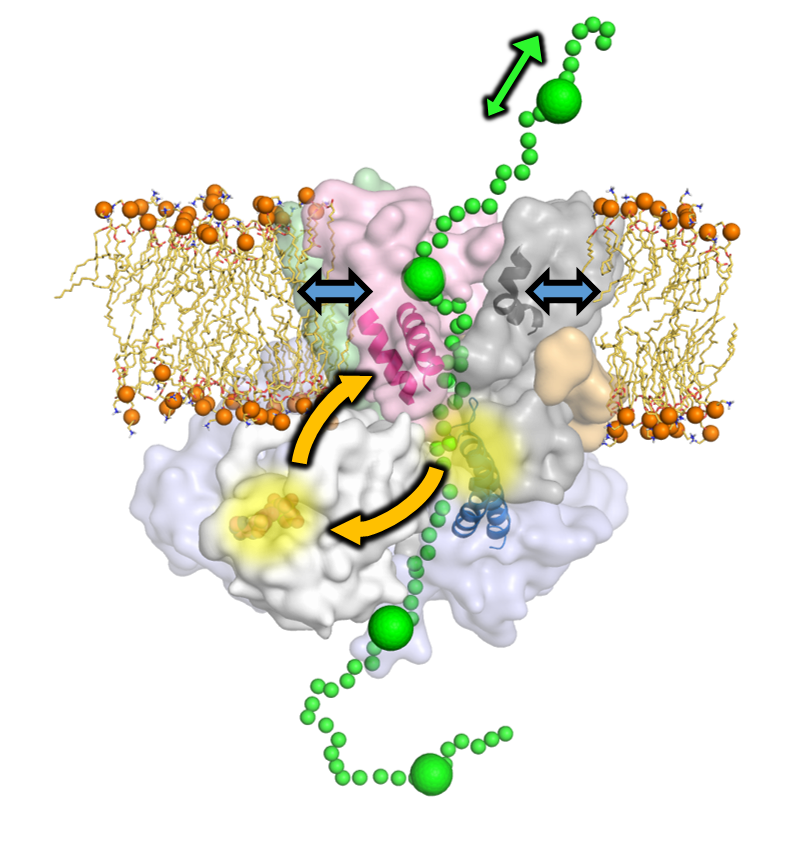
The mechanism of protein translocation across cell membranes
The essential process of protein secretion is achieved by the ubiquitous Sec machinery. In prokaryotes, the drive for translocation through the SecYEG channel comes primarily from ATP hydrolysis by the cytosolic motor-protein SecA. However, the mechanism through which ATP hydrolysis is coupled to directional movement through SecYEG is unclear. Through a combination of all-atom MD simulations and single molecule FRET and biochemical assays, it is shown that ATP binding by SecA causes opening of the SecY-channel at long range, while substrates at the SecY-channel entrance feed back to regulate nucleotide exchange in SecA. This two-way communication suggests a new 'Brownian ratchet' mechanism, whereby ATP binding and hydrolysis bias the direction of polypeptide diffusion. The model represents a solution to the problem of transporting inherently variable substrates such as polypeptides, and may underlie mechanisms of other motors that translocate proteins and nucleic acids.
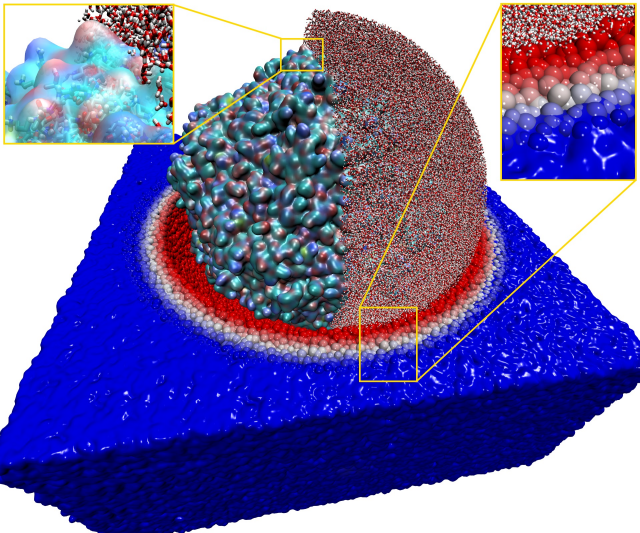
Engineering bacteriophages for treating antimicrobial resistance using all-atom models of entire viruses
The goal of this project was to study an atomistic model of the entire PCV2 virus using molecular dynamics simulation. The developed model has been used to understand how alterations to the structure of the virus enhance its potential AMR (antimicrobial resistance) properties as phage therapy agents.
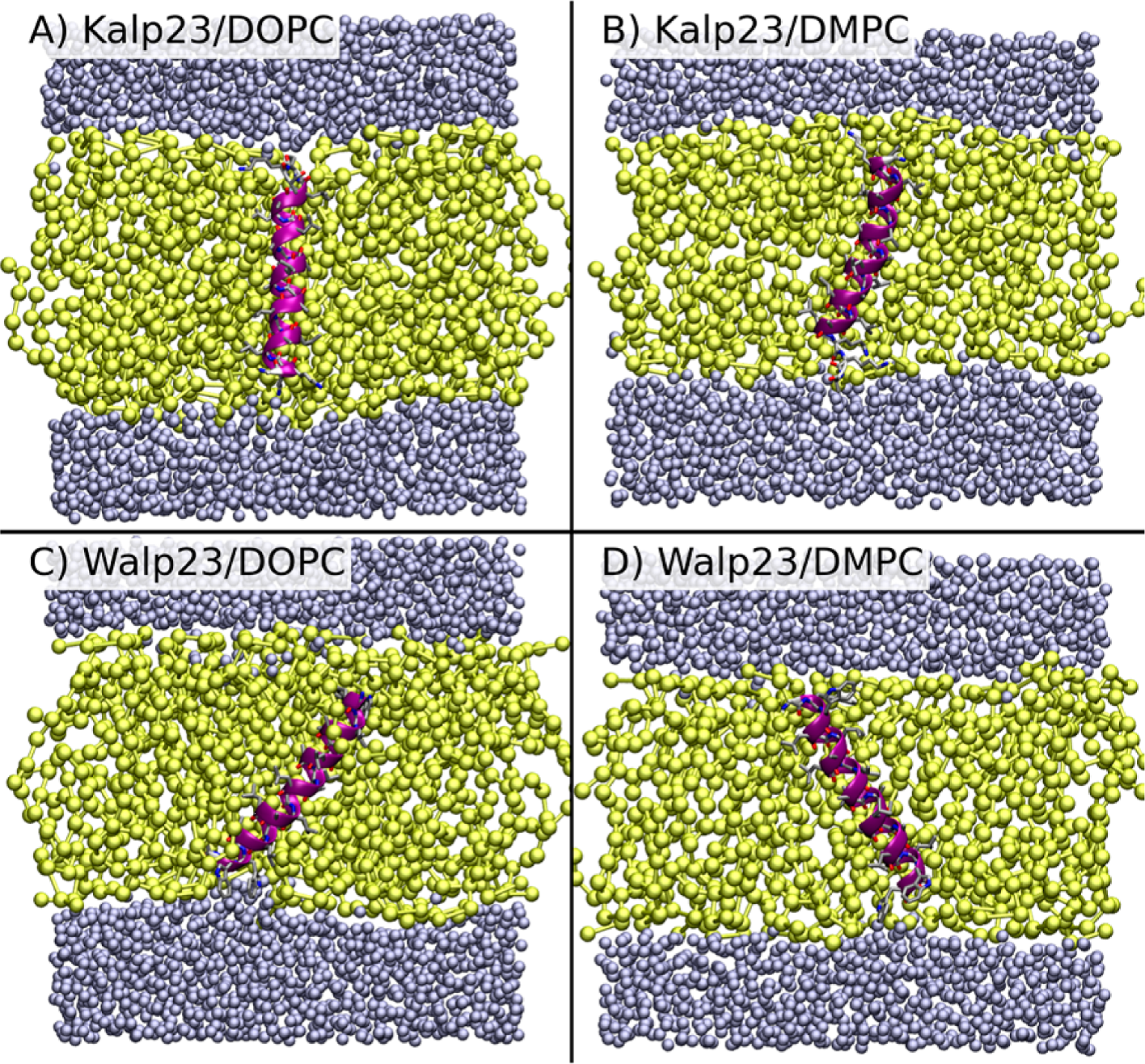
Development of hybrid coarse-grain/atomistic simulation models and their application to membrane-bound proteins
The membrane environment is really important. It acts as a barrier to maintain the integrity of cells, and many important biological processes take place in the membrane, including the requirement of drugs to pass through to reach their binding sites. Computer modelling of membrane systems is therefore crucial to obtaining a full understanding. However, simulating all the atoms, while generally accurate, is expensive to perform owing to the large number of interaction sites. A common method to improve computational efficiency is to reduce the number of interaction sites by subsuming groups of atoms into larger beads. However, this simplification leads to a loss of accuracy. A solution to this problem is to keep part of the system at the atomistic level, and to use a hybrid coarse-grain/atomistic approach. In this project we have developed and tested just such an approach, using it to explore proteins in biological membranes, and how readily small molecules permeate, a critical component of the drug delivery process.
Software Development
Below are a list of eCSE projects (ongoing and completed) involving consortium members, or codes which are used by the consortium:
- eCSE05-10 PI: Dr Oliver O Henrich Adding Multiscale Models of DNA to LAMMPS (12 months)
- eCSE08-14 PI: Dr Thomas W. Keal Task-Farming Parallelisation of Python-ChemShell for Nanomaterials (14 months)
- eCSE08-20 PI: Mr Iain A Bethune Scalable and portable molecular integration with the MIST library (9 months)