


UK Car-Parrinello Consortium, (UKCP)
The UKCP consortium is focused on the application of quantum mechanics to understand and predict the properties of materials. The members of UKCP have made major contributions to both the theory and application of quantum mechanics-based materials modelling, and have developed a common code-base of 3 primary packages (CASTEP, ONETEP and CONQUEST) which are distributed world-wide and widely used in both academia and industry. The consortium contains academics from different disciplines (mainly Physics, Chemistry and Materials) and is still actively developing new methods and algorithms, and pioneering new abilities to calculate even more properties, with high accuracy and efficiency.
| Website | http://www.ukcp.ac.uk/ |
| Contact | |
| Consortium Head | Dr Matt Probert |
| ARCHER CSE Consortium Contact | Dr Toni Collis |

Beating the Stoner criterion using molecular interfaces
Two common metals that are not magnetic - copper and manganese - can be transformed into magnets: a surprising effect that involves combining thin films of the metals with fullerene molecules. Density functional theory simulations suggest a mechanism based on magnetic hardening of the metal atoms, owing to electron transfer. The mechanism may allow for the design of magnetic materials and interfaces using abundant, non-toxic components such as organic semiconductors, with new possibilities for electronic, power or computing applications.
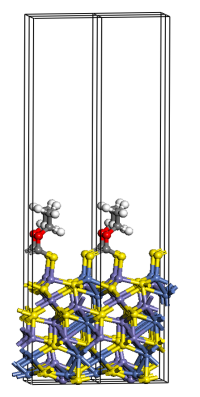
A first principles Study on Ligand Binding and Positional Disorder in Pentlandite
Density functional theory, in conjunction with a cluster expansion model, has been used to study the structure and stability of the positionally disordered iron-nickel sulfide mineral pentlandite (Pn), (Fe, Ni)9S8, with results indicating heterogeneous nearest neighbour metal contacts are more energetically favourable than homogeneous contacts. We also addressed the binding of ethyl xanthate, water and hydroxide to the [111] Pn surface to better understand the mode of action of flotation agents for the recovery of metals by industrial mining processes. In order to model anionic ligands bound to a periodic boundary condition surface we propose a correction scheme derived from the surface work function to remove the additional charge introduced by the ligand. The modelling study was extended to other mineral surfaces and other industrial collector ligands, and a hierarchy of ligand-mineral surface binding emerged, which was subsequently verified by experiment based on electrochemistry measurements.

Ultra-high temperature properties of ZrC: a fully-anharmonic ab-initio approach
Recent advances in finite temperature first-principles methodology have been applied to calculate ultra-high temperature thermodynamic properties of ZrC. These calculations, which represent a first application of such methods within the burgeoning field of ultra-high temperature ceramics (UHTCs), provide fully anharmonic, DFT-level results at temperatures where experimental data is unavailable or inherently unreliable. This data will in future be used within a CALPHAD reassessment of the ZrC phase diagram, leading to an improved understanding of the phase-stability of this material; crucial for understanding its behaviour for applications in, e.g., thermal barriers, also in fission and fusion-reactors.
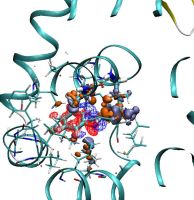
Density functional theory calculations on entire proteins for free energies of binding: Application to a model polar binding site
In drug optimization calculations, the molecular mechanics Poisson-Boltzmann surface area (MMPBSA) method can be used to compute free energies of binding of ligands to proteins. The method involves the evaluation of the energy of configurations in an implicit solvent model. One source of errors is the force field used, which can potentially lead to large errors due to the restrictions in accuracy imposed by its empirical nature. To assess the effect of the force field on the calculation of binding energies, in this project we used large-scale density functional theory (DFT) calculations as an alternative method to evaluate the energies of the configurations in a “QM-PBSA” approach. Our DFT calculations were performed with a near-complete basis set and a minimal parameter implicit solvent model, within the self-consistent calculation, using the ONETEP program on protein–ligand complexes containing more than 2600 atoms. We applied this approach to the T4-lysozyme double mutant L99A/M102Q protein, which is a well-studied model of a polar binding site, using a set of eight small aromatic ligands. We observed that there is very good correlation between the MM and QM binding energies in vacuum but less so in the solvent. The relative binding free energies from DFT are more accurate than the ones from the MM calculations, and give markedly better agreement with experiment for six of the eight ligands. Furthermore, in contrast to MM-PBSA, QMPBSA is able to correctly predict a nonbinder
Software Development
Below are a list of eCSE projects (ongoing and completed) involving consortium members, or codes which are used by the consortium:
- eCSE06-6 PI: Mr Iain A Bethune CP2K - scalable Density Functional Theory (12 months)
- eCSE07-6 PI: Prof. Chris-Kriton Skylaris Implementation and optimisation of advanced solvent modelling functionality in CASTEP and ONETEP (12 months)
- eCSE08-9 PI: Lev Kantorovich CP2K --- Electron Transport based on Non-Equilibrium-Green‰ŕ¤żŕ¤‹s-Functions Method (12 months)
- eCSE08-10 PI: Phil Hasnip Optimal parallelisation in CASTEP (6 months)
- eCSE08-15 PI: Dr Jose Maria Escartin Esteban Implementation of Spin-Orbit Couplings in Linear-Scaling Density Functional Theory (12 months)
- eCSE10-10 PI: Dominik Jochym Enhancing long-range dispersion interaction functionality of CASTEP (8 months)
- eCSE11-17 PI: Phil Hasnip University of York Optimising CASTEP on Intel's Knight's Landing Platform (6 months)