


Pushing the Frontiers of Kinetic Monte Carlo Simulation in Catalysis - Zacros Software Package Development
eCSE001-001Key Personnel
PI/Co-I: Dr Michail Stamatakis, Dr James Hetherington - UCL
Technical: Dr Jens H Nielsen - UCL
Relevant Documents
Technical Report: Zacros Software Package Development: Pushing the Frontiers of Kinetic Monte Carlo Simulation in Catalysis
Project summary
The importance of heterogeneous catalysis in applications that enhance the quality of life cannot be overstated. Iron-based catalysts for the production of ammonia by the Haber process is a notable example, the importance of which is shown by the fact that fertilisers generated from ammonia produced in this way are responsible for sustaining one third of the Earth's population.
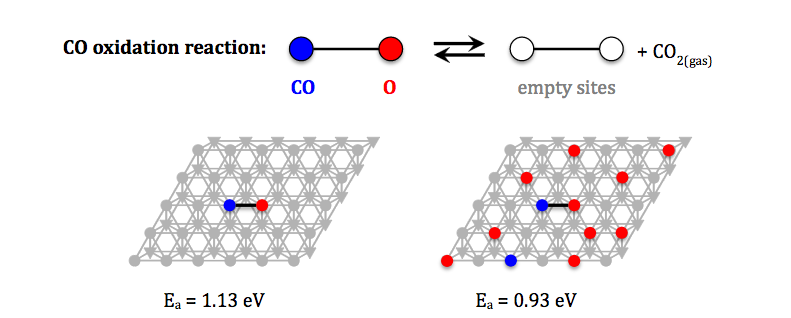
In such catalytic processes, reactant molecules adsorb on the surface of a material (such as a transition metal), and interact with each other and with the catalyst in such a way that: (i) the overall reaction (e.g. ammonia production) is accelerated, and (ii) the material regenerates in the end of the catalytic cycle. However, finding such catalytically active materials is non-trivial: traditional practice involves trial-and-error experimentation, which is time-consuming, costly, and does not guarantee success. On the other hand, simulations can yield vital information and a detailed understanding of how catalysts work and what affects their performance. Our kinetic Monte Carlo (KMC) methodology and our software package Zacros have already contributed to the unravelling of complex phenomena on catalytic surfaces. This knowledge can be used to improve existing catalysts or design novel materials with optimal activity and selectivity for bespoke applications.
However, the computational times required to simulate large domains have limited the accuracy of the predictions and the applicability of the method. For instance, dynamic changes in the surface structure of the catalyst (a phenomenon termed study catalyst reconstruction) have been observed to occur over length scales of hundreds of micrometres, whereas current KMC approaches are limited to domain sizes of tens of nanometres.
In this project, we have made it possible for multiple computers to work together to run simulations of large KMC domains, with each computer tackling a different part of the domain, producing results much more quickly. With the probabilistic nature of statistical mechanics, achieving consistent results between these computers requires careful bookkeeping. We have therefore proceeded by first implementing a solution which is known to be consistent, but where the cooperating computers ensure that only one is acting at a time. This doesn't result in faster simulations, but allows us to prove that our approach to sharing out the work is correct. From this baseline, we can then subtly relax the requirements for consistency, producing approximately correct results much more quickly. This has resulted in 23x speedup using 24 ARCHER nodes. Future work will enable us to exploit an ever-increasing number of machines.
Summary of the software
Zacros is a Kinetic Monte Carlo software package written in Fortran for simulating molecular phenomena on catalytic surfaces. The tool enables researchers in the areas of Computational Catalysis and Surface Science to perform dynamic modelling of adsorption, desorption, surface diffusion, and reaction processes on heterogeneous catalysts. The rates of these elementary processes are typically computed from ab initio simulations, thereby enabling the prediction of catalytic performance metrics (such as activity and selectivity) from first principles, and the detailed validation of hypothesised kinetic mechanisms against experimental data.
The package employs the Graph-Theoretical KMC methodology coupled with cluster expansion Hamiltonians and Brønsted-Evans-Polanyi relations, which can naturally capture:
- steric exclusion effects for species that bind in more than one catalytic site,
- complex reaction patterns involving adsorbates in specific binding configurations and neighbouring patterns,
- spatial correlations and ordering arising from adsorbate lateral interactions that involve many-body contributions,
- changes in the activation energies of elementary events, influenced by the energetic interactions of reactants with neighbouring spectator species.
The software was commercialised by UCL Business in Nov-2013, and has a dedicated website (http://zacros.org) with documentation, tutorials, list of publications and links to e-Lucid, the UCL online licensing portal from which users can obtain Zacros. The code can be obtained free of charge by academics, and since inception it has been distributed to 68 users in 23 countries worldwide.
While not available as a module, the software can be simply installed on ARCHER using the CMAKE based installation tools we have developed and maintain. Requests for access to the Zacros repository on Github are reviewed and granted on an individual basis.