


QuasiParticle Self-Consistent GW electronic structure calculations of many-atom systems
eCSE01-005Key Personnel
PI/Co-I: Prof Mark van Schilfgaarde - King's College London; Dr Martin Lueders - Scientific Computing Department, Science and Technology Facilities Council (STFC)
Technical: Dr Martin Lueders - Scientific Computing Department, Science and Technology Facilities Council (STFC)
Relevant Documents
eCSE Technical Report: QuasiParticle Self-Consistent GW calculations of many-atom systems
Project summary
Many aspects of modern technology require materials with fine-tuned properties. Examples are materials for electronic devices, or materials for regenerative energy production. While the standard model of materials simulations (density functional theory, DFT) is appropriate for a number of applications, there are many cases, such as the calculation of photovoltaic materials and their efficiencies, which are beyond its capability. By construction, DFT is a method to calculate ground state properties of materials, but is not suitable for the calculation of excitation energies, such as the calculation of the absorption spectrum of photovoltaic materials.
A novel method, which is aimed at such excitation energies, has been developed by Kotani, Schilfgaarde et al. over the past years. The new method, called the Quasiparticle Self-Consistent GW approximation (QSGW), is based on many-body perturbation theory. It directly calculates the so-called Green's function of a system, which contains the full information about the excitation spectra. This method is far superior to DFT, but carries a significantly increased computational cost.
A recent APL Materials paper on CH3NH3PbI3 (also known as MAPI) provides an excellent illustration of the power and utility of this method. The original code took about 3 months (on an 8-core machine) for the calculation of a moderately sized system such as MAPI in an idealised crystal structure with 12 atoms. For a quantitative analysis of the recombination losses so crucial in solar cells, it was essential that a high-quality theory such as QSGW be accessible. Through algorithmic improvements (OMP parallelisation and rebundling of loops to take advantage of level 3 BLAS calls where possible) we were able to reduce the time to about 2 days on a 16-core machine.
The paper used an idealized structure of 12 atoms. Had it been possible, a 96-atom structure would have been preferred because of continuous flexing of the PbI3 cages. But even with optimised OMP implementation, calculations in a more realistic structure of this type are beyond reach. To accomplish this, a large-scale machine with memory distribution of key quantities (e.g. matrix elements and screened coulomb interaction) is essential.
The community has acknowledged the importance of this novel QSGW method - it is now considered the gold standard for weakly-correlated materials. It is the current flagship project of the Computational Collaborative Project on Electronic Structure Calculations (CCP9). The main emphasis of the flagship project is the development of new functionality, and the improvement of the user-friendliness of the code. Aspects of the performance have been addressed in a previous d-CSE project on HECToR, and in this eCSE project.
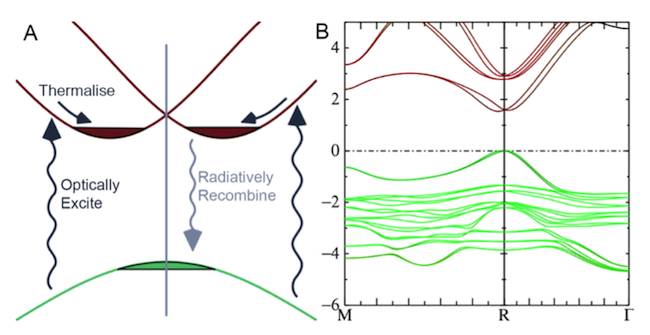
Schematic diagram showing the dual direct-gap/indirect-gap nature of CH3NH3PbI3 (also known as MAPI), a relatively new solar cell material that is both efficient and inexpensive
Summary of the software
The QSGW code is based on the Questaal package, which is a general purpose, linearized muffin-tin orbital method. The codes are held in a git repository on bitbucket (https://bitbucket.org/lmto/). The whole package is also the object of the current flagship software development project of CCP9, a collaborative computational project on first principles electronic structure calculations of condensed matter systems. One important component of the CCP9 flagship project is to make it user-friendly; it is in the process of being designed as a community code to reach a wide user base. There is a web site at https://www.questaal.org/. This eCSE project aimed at the parallelism of the code. Once the CCP9 project is completed the code will be made available as a module on Archer.